
Introduction:
Prion diseases, also known as transmissional spongiform encephalopathy (TSEs),
are a rare category of neurodegenerative diseases that are known for their unusually long incubation periods
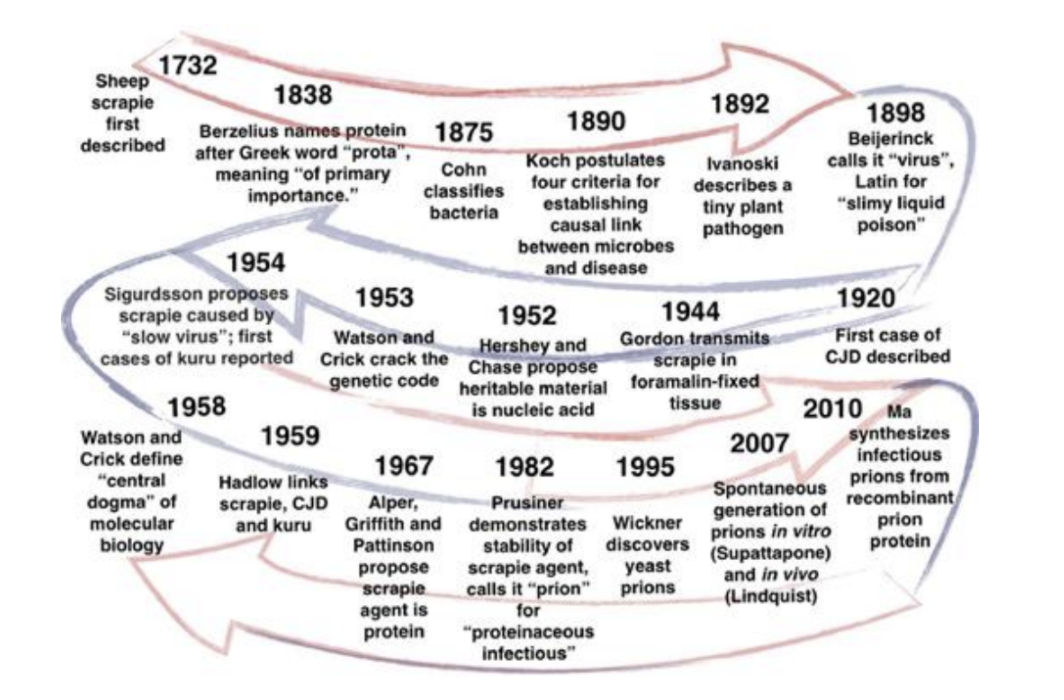
and lethality. While the discovery of TSEs began in the early 18th century with the discovery of scrapies in
sheep, they are still not completely understood and have been subject to extensive research over the last
decade.
However, recent breakthroughs in the understanding of TSEs have opened the way for the
development of potential therapeutics.
The Origin of TSEs:
Unlike bacteria and viruses, which have structures oriented around infecting hosts, prions are simple
proteins that originate throughout the body, including the brain. This protein, referred to as PrP-Sen or
PrP-C, is located specifically in the membrane of nervous cells. Due to reasons that are not fully
understood yet, this protein will sometimes misfold into an alternate form known as PrP-Res or
PrP-Sc.
There are a few differences between these two forms, most importantly being that PrP-Sc contains
significantly more amino acids in the beta helix than its normal counterpart. Another characteristic of
PrP-Sc is its ability to resist protease, which is an enzyme that breaks down misshapen proteins without an
immune response. When a PrP-Sc comes into contact with a normal PrP-C, it causes it to become
misfolded
as well, creating a cascading effect that rapidly transforms the proteins. The PrP-Sc proteins end up
sticking together in long strands attached to neurons known as amyloid fibers. When astrocytes from the
immune system attempt to remove these protein strands, they kill the neuron that they are attached to as
well. In several months, a patient’s brain begins to develop holes, leading to the reasoning for the
“spongiform” in TSEs. While prion diseases come in many different forms affecting both animals and humans,
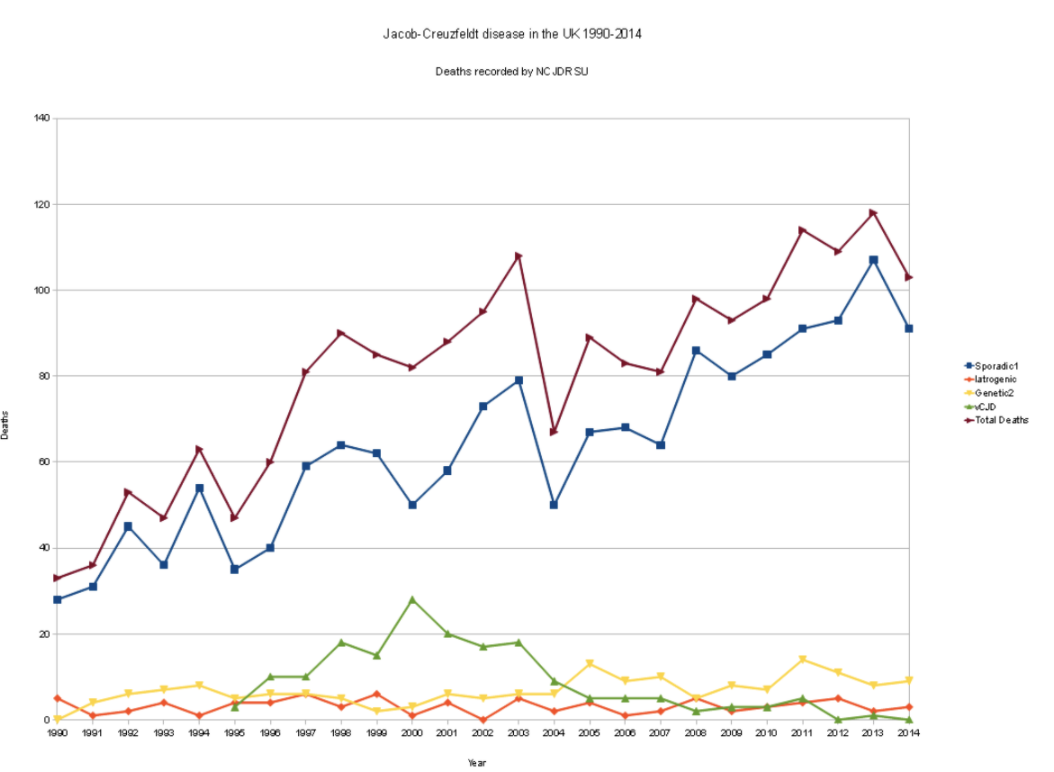
all forms are incredibly rare to contract. The normal form of Creutzfeldt-Jakob Disease (CJD) is the most
common form of prion disease and has been known to be contracted from a variety of sources. Bovine
Spongiform Encephalopathy (BSE) is a prion disease that affects cows and had a large outbreak in northern
England in the mid eighties. If nervous tissue is consumed from infected cattle, a person could contract
variant Creutzfeldt-Jakob Disease (vCJD). If someone contracts the disease through contaminated medical
equipment, medical tissue, or growth hormones, it is known as Iatrogenic Creutzfeldt-Jakob Disease (iCJD).
While it can sometimes be determined where an infection originated
from, more than 85% of cases have an
unknown origin.

Diagnosis And Preventing Spread?
In the case of CJD, It can often be incredibly difficult to determine if someone has actually contracted
the disease. The only way to confirm a diagnosis is through a sample of brain tissue during an autopsy.
However, several tests can be done to rule out any other possible illnesses. Spinal taps and MRI scans are
commonly used in the examination progress. Because it is believed that TSEs can be spread
through
contaminated medical equipment, surgical equipment used in examinations must be deeply cleaned.
The fact that the disease exists in individual proteins makes it much more difficult to disinfect than
bacteria or viruses. Current methods for killing prions require a process of heating equipment to 250
degrees Fahrenheit, applying 21 PSI, using NaOH disinfectant, and letting it sit for an hour. Another
challenge for preventing the spread is with animal forms of TSEs. In the mid-eighties, when BSE had an
outbreak in England, cattle imports from the UK were quickly banned by many countries, including the US.
vCJD was discovered to be caused by ingesting nervous matter from BSE infected cows, and government
institutions had to react quickly in order to prevent future outbreaks. During this time, the USDA
also
created a surveillance program in order to detect cases of BSE before they break out as they had
done in England.

Pathogenicity:
While extensive research has occurred on TSEs since the nineties, lots are still not known about the nature of prions. However, contemporary research has provided a much larger insight into understanding the disease. Recently, researchers from Imperial College London and the University of Zurich were able to determine how PrP-C proteins become misfolded in many sporadic cases of TSEs. By using nuclear resonance spectroscopy and computer simulations, they were able to determine the molecular process in which the protein would misfold. In their study, they were also able to produce antibodies that would prevent the mechanism. However, the antibodies developed currently are too large to pass through the brain, but it has opened the door for therapeutics to potentially be developed in the future. They hope that through their research, drug researchers can create new compounds that could target the mechanism and pass through the brain. One challenge that comes when determining how TSEs spread is the illness’s incredible long incubation period. In acquired forms of the disease, symptoms can take anywhere from a year to several decades to manifest after the initial contact. And because the disease can only be confirmed during the autopsy, discovering what exactly caused the infection can be difficult. Before the late-eighties, surgically injected growth hormones for people had used hormones from adult cows. Once the practice stopped, some people who had taken the hormones had become infected in the following years. However, cases have continued to this day, showing that this form iCJD has an incubation period of nearly 40 years.
Significance
Although TSEs are incredibly rare, understanding the nature of their spread is not only important for helping those who have it, but also for those who suffer from similar neurodegenerative diseases and improving public health. Although it has just remained a theory, some medical professionals believe that prion diseases and Alzheimer's disease are related. One reason for this connection that researchers have pointed out is that both illnesses are triggered by the development of Amyloid fibers in the brain and spinal cord. Through a better understanding of the causes of Prion diseases, drug researchers and pharma companies will be able to provide new therapeutics and medicines for a variety of neurological diseases.
Citations:
Jackson, Christina. Researchers Identify What Causes Prions to Become Pathogenic. 17 Mar.
2021,
www.genengnews.com/news/researchers-identify-what-causes-prions-to-become-pathogenic/.
Zabel, Mark D, and Crystal Reid. “A brief history of prions.” Pathogens and disease vol. 73,9
(2015):
ftv087. doi:10.1093/femspd/ftv087 https://pubmed.ncbi.nlm.nih.gov/26449713/
“Prion Diseases.” National Institute of Allergy and Infectious Diseases, U.S. Department of Health
and
Human Services, www.niaid.nih.gov/diseases-conditions/prion-diseases.
“Prion Diseases.” Centers for Disease Control and Prevention, Centers for Disease Control
and
Prevention, 9 Oct. 2018, www.cdc.gov/prions/index.html.
Cameron, David. “Unweaving Amyloid Fibers to Solve Prion Puzzles.” Whitehead Institute of MIT, 8
June
2005, wi.mit.edu/news/unweaving-amyloid-fibers-solve-prion-puzzles.
“Prion Diseases.” Johns Hopkins Medicine, Johns Hopkins
Medicine,
www.hopkinsmedicine.org/health/conditions-and-diseases/prion-diseases.
Rudge, Peter et al. “Iatrogenic CJD due to pituitary-derived growth hormone with genetically
determined
incubation times of up to 40 years.” Brain : a journal of neurology vol. 138,Pt 11 (2015):
3386-99.
doi:10.1093/brain/awv235 https://pubmed.ncbi.nlm.nih.gov/26268531/